

处方前研究的目的和重要性
处方前研究的目的是提供处方工艺设计的依据,并对处方工艺研究中出现的问题提供机理明确的针对性的解决方案。
1确定粒径是否对溶出度有影响
· BCS II和IV类药物;
· 公开文献显示原研控制了粒径;
· 经验性地判断,常用剂量下,API水中溶解度在100 ug/mL以下时粒径对溶出可能有较显著的影响;
· 制剂开发过程中观察到粒径对溶出有影响
2确定原料药粒径的控制范围
案例1某速释片剂
· 利用多个研发和临床试验批次上获取的数据进行统计回归;
粒径分布D90控制范围的确定:
· 制剂终产品的质量特性在很大程度上是由原料药的性质决定的:尽管也有很多手段来改善制剂的质量特性和工艺可靠性,但这些手段的有效性往往不如对原料药的特性进行控制;
· 现代产品开发的趋势是把控制点前移;
· 在QbD研发要求里,处方前研究的另一重要目的是为制剂研究提供初步风险评估的依据,确定制剂研究的方向和范围;
·
处方前研究的主要内容
处方前研究的主要内容是搞清楚原料药和辅料的理化,生物学,和机械性质。
· 原料药的物理性质:溶解度,晶型,粒径,晶癖等
· 原料药的化学性质:稳定性,辅料相容性等
· 原料药的生物学性质:渗透性,BCS分类,酶代谢稳定性
· 原料药的机械性质:流动性,可压性(塑性和弹性),密度等
处方前研究还需要与制剂研究相结合,由此确定原料药的关键属性CQA:原料药性质对制剂的影响既是制剂研究的工作,也是处方前研究的工作 。
一般NDA申报的原料药理化性质
· SOLID STATE PROPERTIES
m.p., pKa , logD
· POLYMORPHS
· SOLUBILITY AND PERMEABILITY
· WATER CONTENT (hygroscopicity)
· CHEMICAL AND PHYSICAL STABILITY
· PARTICLE SIZE, SURFACE AREA, AND DENSITY
· MECHANICAL PROPERTIES:
PLASTICITY, ELASTICITY,
COMPRESSIBILITY, FLOWABILITY
考察的项目需要考虑一般性质和与该原料和制剂相关的性质。
仿制药的晶型选择:理解原研药的晶型(物理状态)的选择原则
· 一般情况下原研药品已经做了晶型的筛选。
· 原研药的多晶型筛选的基本目的是确定并选择最稳态晶型,以降低晶型突变的风险,而不是改善原料药性质;
· 结晶优于无定型,热力学最稳定晶型优于亚稳态晶型势能态越低越稳定;高势能态有转化为低势能态的趋势,尽管动力学速率可能很慢。
· 无定型的选择一般只在两种情况下:原料药找不到结晶态或生物利用度太低且没有其它更好的方法加以提高;
· 水合物与无水物之间的选择主要看哪一个在药品储存条件下更稳定及相互转化的条件;
· 平衡与取舍难以避免;有时不得不选择不常用的或不够稳定的物理状态。
1原料药的理化性质受其物理状态影响
常见固体物理状态的结构差别

结构有序程度:最稳态结晶 =亚稳态结晶>液晶>无定型
2常见固体物理状态的能量差别
· 能量(自由能)差别:最稳态结晶<亚稳态结晶 <液晶 <无定型;
粒径和溶出是否要分别独立控制?例如文献报导了原研的粒径范围,是否溶出和粒径都需要与原研一致?
· 在粒径对溶出有显著影响的情况下,粒径和溶出的控制往往不可能相互独立。溶出是目标,粒径控制是手段;
· 文献报道原研粒径的控制范围时通常不注明测定方法和仪器,因此数值不一定能直接转移;
· 有的情况下,溶出(包括常用的四个条件)与原研吻合,但仍不BE。研究者可能因此考虑进一步调整粒径到原研的控制范围。这里药学上可以先考虑溶出方法的体内外相关性,试验更多的溶出方法;同时考虑药代特性,如体内变异的情况来查找原因
化学稳定性
处方前的研究内容:
· 原料药的化学结构和活性基团;
· 降解机理,途径,和产物:水解,氧化,聚合,光解等;
· 稳定性影响因素(强制降解实验):pH、水分、温度、光照等;对pH不稳定的药物需要研究pH-stability曲线以确定最稳定范围;
· 稳定性影响因素研究的结果为处方工艺包材的选择,以及制剂研发中出现的质量问题提供机理性的解决方案。
1辅料和包材的化学性质常被忽视
· 建立辅料包材数据库:收集辅料包材的易反应基团,pH、过氧化物、金属离子、浸出物等杂质信息,指导针对性地选择辅料和工艺;
· 原辅料包材相容性试验:验证辅料包材的选择结果。
案例1苯磺酸氨氯地平片
文献1: 苯磺酸氨氯地平片的制备工艺研究,郑敏 - 《内蒙古医学杂志》,2012, 44(9)
【摘要】:…处方为:苯磺酸氨氯地平6.93g、微晶纤维素80g、乳糖80g、低取代羟丙基纤维素(L-HPC)9g、5%淀粉浆适量、硬脂酸镁1.5g。制备工艺为:将处方中的原辅料皆粉碎过80目筛,称取处方量的苯磺酸氨氯地平、微晶纤维素、乳糖、低取代羟丙基纤维素,先将除苯磺酸氨氯地平及硬脂酸镁外的其它辅料混匀,按照等量递增法将苯磺酸氨氯地平与混合好的辅料混匀,加5%淀粉浆制成软材,过20目筛制粒,50~60℃烘干,18目筛整粒,加硬脂酸镁,混合均匀,测颗粒含量,计算片重,7.5mm浅凹冲压片,即得结论:本品处方设计合理,工艺稳定可行。
文献2: HPLC法检测苯磺酸氨氯地平片剂中的乳糖相关杂质,吴暎 刘伟 林萍 –《药学进展》 2013年02期
【摘要】:…结论:该法可用于测定苯磺酸氨氯地平片剂中的乳糖相关杂质含量…【正文快照】: 苯磺酸氨氯地平为…在苯磺酸氨氯地平片剂的处方研究中,笔者发现制剂中有一种随着放置时间延长而含量明显增加的杂质(命名为杂质Ⅰ,2),而苯磺酸氨氯地平原料在同等条件下无此杂质…
点评:
· 氨醛缩合(Millard reaction)反应是制剂处方中较常见的相容性问题;
· 一个教科书上已讨论过的问题为什么还会反复出现?
对药物的结构和活性集团不够重视,处方设计的时候没有考虑
案例2兰索拉唑肠溶片
文献1:兰索拉唑肠溶片稳定性和溶出度的影响因素研究 ,王贺,孙备,李姜晖,刘羽(安徽省药物研究所),中国药业,2010,19(14),32
粒径和溶出是否要分别独立控制?例如文献报导了原研的粒径范围,是否溶出和粒径都需要与原研一致?
点评:
· 影响因素考察没有研究基本理化参数如pH的影响,而是直接选用不同的碱来试验;因而没能确定药物的最佳稳定性pH区间及碱的pH和组成;
· 由于上述缺陷,对关键辅料无法实现有效控制,当辅料变更的时候,问题又会出现。
案例3某仿制冻干粉针
背景资料:
· APIpka:6.0
· 原研标签pH范围:3.0-4.5
· 原研稳定性:25±2°C/60±5% RH可稳定18个月,40±2°C/75±5% RH加速条件可稳定6个月
· 吸湿性:无吸湿性
· 溶解度:溶于水和0.9%氯化钠溶液
PH调节范围的选择
· 在粒径对溶出有显著影响的情况下,粒径和溶出的控制往往不可能相互独立。溶出是目标,粒径控制是手段;
· 文献报道原研粒径的控制范围时通常不注明测定方法和仪器,因此数值不一定能直接转移;
· 有的情况下,溶出(包括常用的四个条件)与原研吻合,但仍不BE。研究者可能因此考虑进一步调整粒径到原研的控制范围。这里药学上可以先考虑溶出方法的体内外相关性,试验更多的溶出方法;同时考虑药代特性,如体内变异的情况来查找原因
化学稳定性
处方前的研究内容:
· 原料药的化学结构和活性基团;
· 降解机理,途径,和产物:水解,氧化,聚合,光解等;
· 稳定性影响因素(强制降解实验):pH、水分、温度、光照等;对pH不稳定的药物需要研究pH-stability曲线以确定最稳定范围;
· 稳定性影响因素研究的结果为处方工艺包材的选择,以及制剂研发中出现的质量问题提供机理性的解决方案。
1辅料和包材的化学性质常被忽视
· 建立辅料包材数据库:收集辅料包材的易反应基团,pH、过氧化物、金属离子、浸出物等杂质信息,指导针对性地选择辅料和工艺;
· 原辅料包材相容性试验:验证辅料包材的选择结果。
案例1苯磺酸氨氯地平片
文献1: 苯磺酸氨氯地平片的制备工艺研究,郑敏 - 《内蒙古医学杂志》,2012, 44(9)
【摘要】:…处方为:苯磺酸氨氯地平6.93g、微晶纤维素80g、乳糖80g、低取代羟丙基纤维素(L-HPC)9g、5%淀粉浆适量、硬脂酸镁1.5g。制备工艺为:将处方中的原辅料皆粉碎过80目筛,称取处方量的苯磺酸氨氯地平、微晶纤维素、乳糖、低取代羟丙基纤维素,先将除苯磺酸氨氯地平及硬脂酸镁外的其它辅料混匀,按照等量递增法将苯磺酸氨氯地平与混合好的辅料混匀,加5%淀粉浆制成软材,过20目筛制粒,50~60℃烘干,18目筛整粒,加硬脂酸镁,混合均匀,测颗粒含量,计算片重,7.5mm浅凹冲压片,即得结论:本品处方设计合理,工艺稳定可行。
文献2: HPLC法检测苯磺酸氨氯地平片剂中的乳糖相关杂质,吴暎 刘伟 林萍 –《药学进展》 2013年02期
【摘要】:…结论:该法可用于测定苯磺酸氨氯地平片剂中的乳糖相关杂质含量…【正文快照】: 苯磺酸氨氯地平为…在苯磺酸氨氯地平片剂的处方研究中,笔者发现制剂中有一种随着放置时间延长而含量明显增加的杂质(命名为杂质Ⅰ,2),而苯磺酸氨氯地平原料在同等条件下无此杂质…
点评:
· 氨醛缩合(Millard reaction)反应是制剂处方中较常见的相容性问题;
· 一个教科书上已讨论过的问题为什么还会反复出现?
对药物的结构和活性集团不够重视,处方设计的时候没有考虑
案例2兰索拉唑肠溶片
文献1:兰索拉唑肠溶片稳定性和溶出度的影响因素研究 ,王贺,孙备,李姜晖,刘羽(安徽省药物研究所),中国药业,2010,19(14),32
· 能量的差别决定了物理态的溶解度和稳定性:势能越高,溶解度越高,而稳定性越差.。

稳定性: 高自由能的状态将自发转化为低自由能状态,只是动力学速率有快慢之分。
3晶型变更的一般性结论
从上述介绍可以看出:
· 不同的物理形态并非完全等同而可以自由互换;
· 物理形态变更对药品质量的影响并非一定,需要具体评估
·
4晶型变更的法规要求
现有的指导原则包括:
1. Guidance for Industry, ANDAs: Pharmaceutical Solid Polymorphism; July 2007;
2. ICH Q6A Guideline;
3. Guidance for Industry, Regulatory Classification of Pharmaceutical Co-Crystals, Apr 2013
5现行法规对原料药物理状态变更的要求
· 指导原则保留了在仿制药中采用任何物理状态的灵活性,包括亚稳态和无定型;
· 对仿制药,FDA ANDA指导原则提出物理形态的变更需要评估三类可能的影响:对溶解度、溶出度和生物利用度;对产品生产工艺;及对稳定性;
· 对稳定性的要求是产品质量的稳定,而没有直接要求物理状态稳定(状态稳定可能是知识产权的要求),也不鼓励监测制剂中API的物理状态;
· 在ANDA申请时,除上述关于晶型的指导原则外,还需要考虑晶型变更须符合其它指导原则,如FDA关于仿制药的外形尺寸,掰片等指导原则。
·
6仿制药中晶型变更的处理
· 在原研晶型无专利保护的情况下,仿制药应尽量采用原研晶型,尽量避免亚稳态晶型;
· 无定型的结晶倾向大于亚稳态晶型,但可预测性较高;
· 水份的存在会大大加快无定型的结晶速度;
· 如考虑变更,需要评估三类可能的影响:
o 溶解度、溶出度和生物利用度:溶解度较大的药物变更的影响可能不大;溶解度较小的BCSII和IV类药物一般较难变更。
o 稳定性:如果API的稳定性不佳,杂质特别是基因毒性杂质限度很严,则需要认真评估其可能影响。
o 产品生产工艺:一般可以调整工艺来解决。
另外还需要考虑关于仿制药的其它要求。
7亚稳态晶型变更的案例
案例一:稳定性下降,杂质超标;
案例二:密度下降,导致片剂尺寸超出FDA指导原则的要求,对高载药量的产品特别需要注意。
8有关制剂中晶型的常见问题
如何确定原研采用的晶型:
· 掌握原研的晶型选择思路:通常原研采用的一定是最稳态晶型,如在有结晶的情况下不会采用无定型,除非出于增加溶解度的原因;
· 专利和公开文献一般会揭示原研晶型或是否有特殊原因采用了非最稳态;
· 如果文献没有明确晶型,最可靠的是做晶型的筛选和评估确定最稳态晶型;
· 直接检查原研制剂的晶型只在部分载药量较高的情况下才可行,且要确定辅料没有干扰,因此可靠性有限。
如何检查制剂中晶型的稳定性?
· 法规没有要求制剂中晶型一定要稳定,且一般情况下不鼓励直接检查制剂中晶型的稳定性;
· 一般情况下通过检查制剂的质量指标特别是与晶型有关的指标的稳定性来替代(surrogate test);
· 必要时可以通过API在制剂工艺和储存条件下的晶型稳定性来替代:固体制剂辅料对API晶型转变的影响相当罕见;
· 当制剂中有意使用非最稳态晶型且晶型转变对制剂质量指标有影响时,直接检查制剂中晶型的稳定性是有益的,但不是必须的。
·
溶解度
四类溶解度信息:
· pH-溶解度:制剂工艺开发参考
· 溶剂中溶解度:原料药和制剂工艺开发参考
· 溶出介质中溶解度:溶出方法开发参考
生物相关介质中溶解度: SGF, SIF, etc.评估生物药剂学分类参考


案例
1某速释片剂溶出度偏低
· API为强酸弱碱盐
· pKa:2.3
· BCS分类:II类,低溶高渗
· API溶解度受pH影响极大,在3.5以上几乎为零

检查API的性质
· API质量标准中pH范围为0-1.5;
· 实测pH约1.3-1.4;
· 尽管API的pH在质标范围内,但离限度边缘很近,成盐不完全,导致溶出度下降
通常这类pH-溶解度急剧下降的药物同时存在与PPI共用的吸收问题,通过文献检索可以发现;
国内API的pH在质标限度边缘的情况并不少见,常与制剂的溶出度和稳定性问题有关,制剂开发时需要特别注意。
2某速释片剂稳定性试验中溶出度下降
· API为强酸弱碱盐
· pka:3.8
· BCS分类:II类,低溶高渗
问题: 稳定性试验中溶出度下降
检查API性质:
· API:未发现API在稳定性试验中有变化;
· 原辅料相容性: 猜测是API与某个辅料有相互作用;逐个检查后发现可能是与SLS成盐
API与辅料成盐
· 含有SLS的处方比去除SLS的处方溶出更慢;
· 原料药的十二烷基磺酸盐溶解度降低;
措施:处方中弃用SLS

3某速释片剂水中溶出不完全
· API为弱酸弱碱盐
· pka:8.5
· BCS分类:高溶高渗
· 在37℃的水和各溶出介质中的溶解度均大于20mg/ml;
· 问题:片剂水中溶出不完全


API溶解度较高,溶出条件远超Sink condition,为什么溶出不完全?

检查:原辅料相容性
· API为弱碱盐,与处方中交联羧甲基纤维素钠形成不溶性盐;
· 该盐在酸中溶解,而原研的溶出度测定介质是0.1N HCl,因而原研没有发现;
· 上述2,3两个案例可见,稳定性试验中溶出度下降或漏槽条件下溶出不完全往往预示API成盐状态发生变化,可能由原辅料相互作用引起;
通常原辅料相容性试验往往不考察成盐和物理状态的变化,不容易发现此类相互作用
小结:仿制药中溶解度/溶出度问题的常用解决方案
o 原料药的pH-溶解度和BCS分类;
o 原料药盐的控制:API的pH,同时检查辅料的性质,包括pH和pKa;
o 原料药晶型的控制;
o 原料药粒径控制:在第三节中讨论;
o 选择和调整处方,如表面活性剂、崩解剂、水溶性辅料、润滑剂等的用量;
o 选择和调整工艺。
对仿制药来说,由于一致性的要求,一般优先考虑与原研相同的增溶手段
粒径—原研药控制粒径的思路
· CH Q6A决策树提出是否需要控制粒径的程序,需要考察对制剂5个方面的影响:溶出度/生物利用度,稳定性,含量均匀度,外观,生产工艺;
· 对口服固体制剂来说,最常见的是粒径对溶出度/生物利用度的影响;
· BCS II和IV类药物通常都需要控制粒径
不同粒径的体外溶出曲线:

1确定粒径是否对溶出度有影响
· BCS II和IV类药物;
· 公开文献显示原研控制了粒径;
· 经验性地判断,常用剂量下,API水中溶解度在100 ug/mL以下时粒径对溶出可能有较显著的影响;
· 制剂开发过程中观察到粒径对溶出有影响
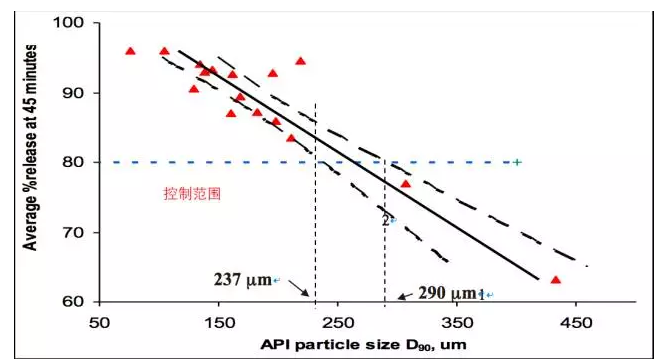
2确定原料药粒径的控制范围
案例1某速释片剂
· 利用多个研发和临床试验批次上获取的数据进行统计回归;
粒径分布D90控制范围的确定:
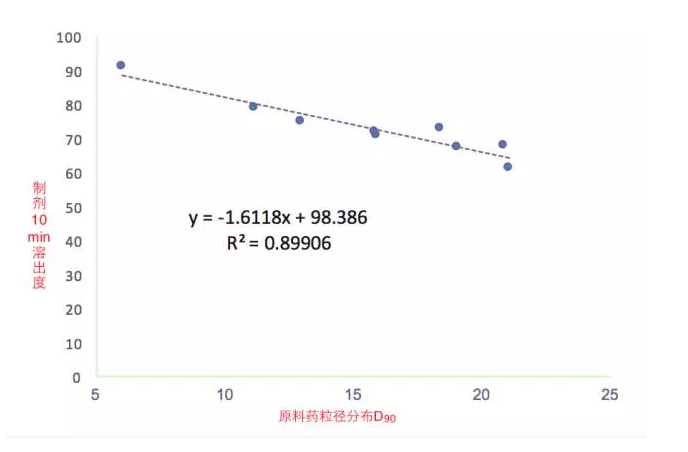
案例2某速释片剂
· 采用类似的回归分析的办法确定粒径控制范围;
· 得到粒径控制范围为12-15um;
· 在粒径控制范围很窄的情况下特别适用。
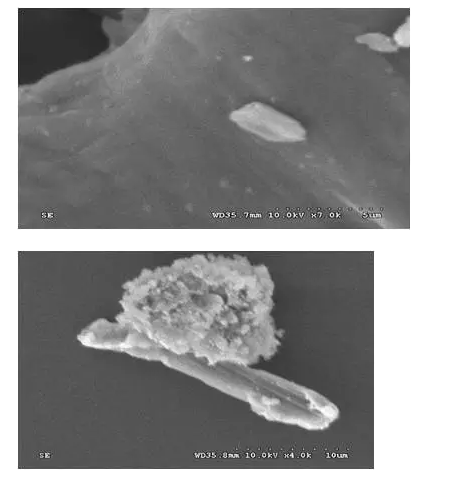
3有关粒径控制的常见问题
可否用显微分析得到原研制剂的粒径?
1. 显微分析得到的粒径数据与光散射不同,数值不能直接转移;
2. 当API结晶为针状片状时,显微观察难以确定粒径大小;
粒径表达为大量颗粒的分布情况,显微观察原研制剂往往难以避开辅料的干扰准确测量大量颗粒;

粒径和溶出是否要分别独立控制?例如文献报导了原研的粒径范围,是否溶出和粒径都需要与原研一致?
· 在粒径对溶出有显著影响的情况下,粒径和溶出的控制往往不可能相互独立。溶出是目标,粒径控制是手段;
· 文献报道原研粒径的控制范围时通常不注明测定方法和仪器,因此数值不一定能直接转移;
· 有的情况下,溶出(包括常用的四个条件)与原研吻合,但仍不BE。研究者可能因此考虑进一步调整粒径到原研的控制范围。这里药学上可以先考虑溶出方法的体内外相关性,试验更多的溶出方法;同时考虑药代特性,如体内变异的情况来查找原因
化学稳定性
处方前的研究内容:
· 原料药的化学结构和活性基团;
· 降解机理,途径,和产物:水解,氧化,聚合,光解等;
· 稳定性影响因素(强制降解实验):pH、水分、温度、光照等;对pH不稳定的药物需要研究pH-stability曲线以确定最稳定范围;
· 稳定性影响因素研究的结果为处方工艺包材的选择,以及制剂研发中出现的质量问题提供机理性的解决方案。
1辅料和包材的化学性质常被忽视
· 建立辅料包材数据库:收集辅料包材的易反应基团,pH、过氧化物、金属离子、浸出物等杂质信息,指导针对性地选择辅料和工艺;
· 原辅料包材相容性试验:验证辅料包材的选择结果。
案例1苯磺酸氨氯地平片
文献1: 苯磺酸氨氯地平片的制备工艺研究,郑敏 - 《内蒙古医学杂志》,2012, 44(9)
【摘要】:…处方为:苯磺酸氨氯地平6.93g、微晶纤维素80g、乳糖80g、低取代羟丙基纤维素(L-HPC)9g、5%淀粉浆适量、硬脂酸镁1.5g。制备工艺为:将处方中的原辅料皆粉碎过80目筛,称取处方量的苯磺酸氨氯地平、微晶纤维素、乳糖、低取代羟丙基纤维素,先将除苯磺酸氨氯地平及硬脂酸镁外的其它辅料混匀,按照等量递增法将苯磺酸氨氯地平与混合好的辅料混匀,加5%淀粉浆制成软材,过20目筛制粒,50~60℃烘干,18目筛整粒,加硬脂酸镁,混合均匀,测颗粒含量,计算片重,7.5mm浅凹冲压片,即得结论:本品处方设计合理,工艺稳定可行。
文献2: HPLC法检测苯磺酸氨氯地平片剂中的乳糖相关杂质,吴暎 刘伟 林萍 –《药学进展》 2013年02期
【摘要】:…结论:该法可用于测定苯磺酸氨氯地平片剂中的乳糖相关杂质含量…【正文快照】: 苯磺酸氨氯地平为…在苯磺酸氨氯地平片剂的处方研究中,笔者发现制剂中有一种随着放置时间延长而含量明显增加的杂质(命名为杂质Ⅰ,2),而苯磺酸氨氯地平原料在同等条件下无此杂质…
点评:
· 氨醛缩合(Millard reaction)反应是制剂处方中较常见的相容性问题;
· 一个教科书上已讨论过的问题为什么还会反复出现?
对药物的结构和活性集团不够重视,处方设计的时候没有考虑
案例2兰索拉唑肠溶片
文献1:兰索拉唑肠溶片稳定性和溶出度的影响因素研究 ,王贺,孙备,李姜晖,刘羽(安徽省药物研究所),中国药业,2010,19(14),32
粒径和溶出是否要分别独立控制?例如文献报导了原研的粒径范围,是否溶出和粒径都需要与原研一致?
· 在粒径对溶出有显著影响的情况下,粒径和溶出的控制往往不可能相互独立。溶出是目标,粒径控制是手段;
· 文献报道原研粒径的控制范围时通常不注明测定方法和仪器,因此数值不一定能直接转移;
· 有的情况下,溶出(包括常用的四个条件)与原研吻合,但仍不BE。研究者可能因此考虑进一步调整粒径到原研的控制范围。这里药学上可以先考虑溶出方法的体内外相关性,试验更多的溶出方法;同时考虑药代特性,如体内变异的情况来查找原因
化学稳定性
处方前的研究内容:
· 原料药的化学结构和活性基团;
· 降解机理,途径,和产物:水解,氧化,聚合,光解等;
· 稳定性影响因素(强制降解实验):pH、水分、温度、光照等;对pH不稳定的药物需要研究pH-stability曲线以确定最稳定范围;
· 稳定性影响因素研究的结果为处方工艺包材的选择,以及制剂研发中出现的质量问题提供机理性的解决方案。
1辅料和包材的化学性质常被忽视
· 建立辅料包材数据库:收集辅料包材的易反应基团,pH、过氧化物、金属离子、浸出物等杂质信息,指导针对性地选择辅料和工艺;
· 原辅料包材相容性试验:验证辅料包材的选择结果。
案例1苯磺酸氨氯地平片
文献1: 苯磺酸氨氯地平片的制备工艺研究,郑敏 - 《内蒙古医学杂志》,2012, 44(9)
【摘要】:…处方为:苯磺酸氨氯地平6.93g、微晶纤维素80g、乳糖80g、低取代羟丙基纤维素(L-HPC)9g、5%淀粉浆适量、硬脂酸镁1.5g。制备工艺为:将处方中的原辅料皆粉碎过80目筛,称取处方量的苯磺酸氨氯地平、微晶纤维素、乳糖、低取代羟丙基纤维素,先将除苯磺酸氨氯地平及硬脂酸镁外的其它辅料混匀,按照等量递增法将苯磺酸氨氯地平与混合好的辅料混匀,加5%淀粉浆制成软材,过20目筛制粒,50~60℃烘干,18目筛整粒,加硬脂酸镁,混合均匀,测颗粒含量,计算片重,7.5mm浅凹冲压片,即得结论:本品处方设计合理,工艺稳定可行。
文献2: HPLC法检测苯磺酸氨氯地平片剂中的乳糖相关杂质,吴暎 刘伟 林萍 –《药学进展》 2013年02期
【摘要】:…结论:该法可用于测定苯磺酸氨氯地平片剂中的乳糖相关杂质含量…【正文快照】: 苯磺酸氨氯地平为…在苯磺酸氨氯地平片剂的处方研究中,笔者发现制剂中有一种随着放置时间延长而含量明显增加的杂质(命名为杂质Ⅰ,2),而苯磺酸氨氯地平原料在同等条件下无此杂质…
点评:
· 氨醛缩合(Millard reaction)反应是制剂处方中较常见的相容性问题;
· 一个教科书上已讨论过的问题为什么还会反复出现?
对药物的结构和活性集团不够重视,处方设计的时候没有考虑
案例2兰索拉唑肠溶片
文献1:兰索拉唑肠溶片稳定性和溶出度的影响因素研究 ,王贺,孙备,李姜晖,刘羽(安徽省药物研究所),中国药业,2010,19(14),32
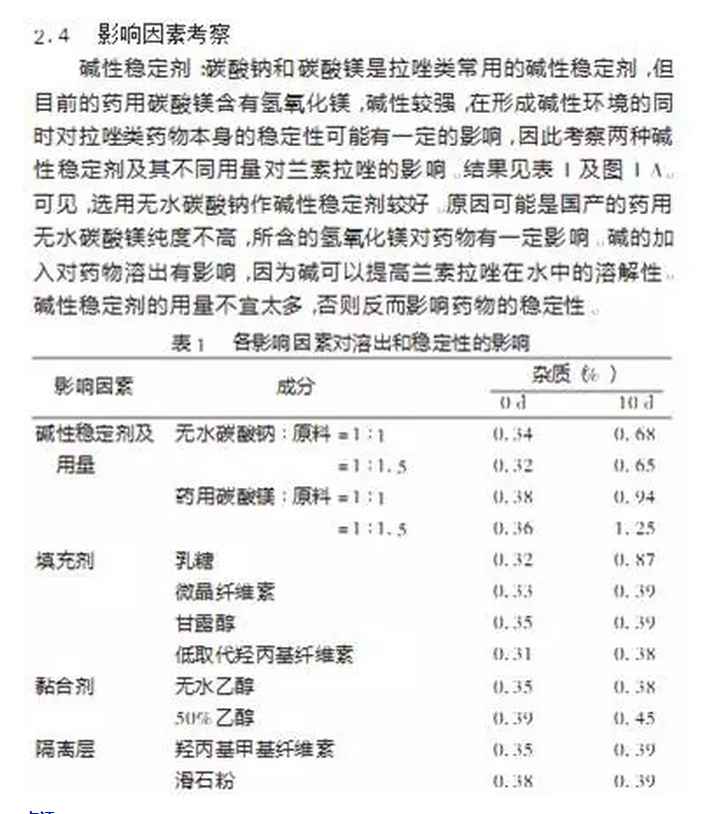
点评:
· 影响因素考察没有研究基本理化参数如pH的影响,而是直接选用不同的碱来试验;因而没能确定药物的最佳稳定性pH区间及碱的pH和组成;
· 由于上述缺陷,对关键辅料无法实现有效控制,当辅料变更的时候,问题又会出现。
案例3某仿制冻干粉针
背景资料:
· APIpka:6.0
· 原研标签pH范围:3.0-4.5
· 原研稳定性:25±2°C/60±5% RH可稳定18个月,40±2°C/75±5% RH加速条件可稳定6个月
· 吸湿性:无吸湿性
· 溶解度:溶于水和0.9%氯化钠溶液
PH调节范围的选择
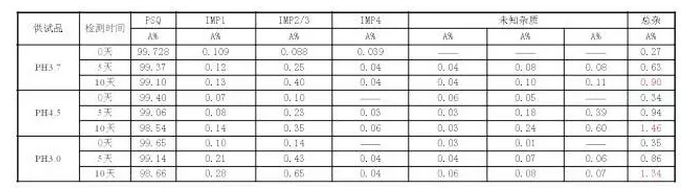
结果显示:在40℃条件下,PH3.7的冻干样品比PH3.0和PH4.5的冻干样品品稳定性好,杂质增长小;结论:确定产品质量标准中PH值范围在3.0-4.5。
点评:
· 由于pH范围中点3.7较边界3.0和4.5显著稳定,需要增加更密集的pH点以确定较窄的控制范围,不能完全照搬原研的标准;
· 不少原研制剂也出现缺陷(这里不详述),主要是由于创新药和仿制药的研发要求不完全相同,原研制剂的缺陷有时在其研发过程中未能发现或及时纠正;仿制时需要评估是否要纠正
小结:
· 制剂质量问题的调整和解决首先考虑原料药的性质,其次是辅料包材的相容性,包括物理相容性;再次考虑制剂的处方工艺;
· 在仿制药的研发中由于可以获得对照品的部分技术信息,研究者往往容易直接参考这些信息而忽视了对问题机理的研究,在出现问题时缺乏可靠的手段加以解决;
从方法学上来说,处方前研究是为制剂研发提供机理性的解决方案,这与DoE方法既有区别又有联系。

上一篇:TOC放行清洁验证的局限性浅析
下一篇:【验证专题】干热灭菌验证怎么做?

 德斯特GMP咨询
德斯特GMP咨询 13427069959
13427069959
